Next Generation Sequencing: Principle, Steps Involved, and Applications
Next Generation Sequencing: Principle, Steps Involved, and Applications
Next Generation Sequencing (NGS) technology has transformed how clinical researchers and scientists think about genetics, as it assesses multiple genes in a single assay. It can sequence an entire or particular genome of interest within a short period. Several different NGS platforms use other sequencing technologies. Still, one common thing is ‘all NGS platforms execute sequencing of millions of small fragments of DNA in parallel.’ Bioinformatics analyses are then used to piece together these fragments.
Principle of Next-Generation Sequencing
Next Generation Sequencing technology is similar to Capillary Electrophoresis(CE) sequencing, where DNA polymerase catalyzes the incorporation of fluorescently labeled deoxyribonucleotide triphosphates (dNTPs) into a DNA template strand during sequential cycles of DNA synthesis. During each process, the nucleotides are identified by fluorophore excitation at the addition of each nucleotide. The major difference is that instead of sequencing a single DNA fragment, NGS simultaneously extends this process across millions of fragments. NGS delivers high accuracy, an elevated yield of error-free readings, and a high percentage of base calls above Q30.
Basic Steps Involved in Next-Generation Sequencing
Illumina sequencer includes four basic steps of sequencing: Library Preparation, Cluster Generation, Sequencing, and Analysis. After the isolation of DNA or cDNA (synthesized from RNA), they undergo all of these four basic steps, which are described below:
The library preparation is vital in the sequencing process as it prepares the samples to be compatible with the sequencer. The samples, either DNA or cDNA, are fragmented by sonication or enzymatic restriction to obtain fragments of 200-500 bp in length. During fragmentation, each fragment gets an overhang A tail at the end, preparing them to ligate to the adapter sequence, which contains a ‘T’- base overhang complementary to the A-tail fragment.
Adapters are the sequence that contains the primer binding sites, index sequences, and the sequence that allows library fragments to attach to the flow cell lawn. These adapters are ligated in 5’ and 3’ ends. Alternatively, a process called tagmentation combines fragmentation and ligation reactions in a single step, increasing the efficiency of the library preparation process.
It helps in the specific enrichment of adapter-ligated DNA during the PCR step.
 Figure: The outer region in black and orange binds to the complementary sequence on the surface of the Illumina flow cell. Through which the individual library single strands are captured to be sequenced. The inner region of green and blue acts as a sequencing primer binding site which is used to read out the insert sequence during the actual sequencing process.Source:https://www.lexogen.com/rna-lexicon-next-generation-sequencing/
Figure: The outer region in black and orange binds to the complementary sequence on the surface of the Illumina flow cell. Through which the individual library single strands are captured to be sequenced. The inner region of green and blue acts as a sequencing primer binding site which is used to read out the insert sequence during the actual sequencing process.Source:https://www.lexogen.com/rna-lexicon-next-generation-sequencing/
For each sample, unique adapters are used, and all the samples are pooled together in a single tube. After pooling together, it is loaded in the sequencer for further proceeding into sequencing.
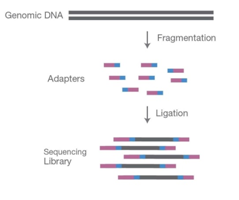 Figure: Fragmenting DNA sample and ligating specialised adapters to both fragments ends to make NGS library. Source:https://www.cd-genomics.com/blog/principle-and-workflow-of-illumina-next-generation-sequencing/
Figure: Fragmenting DNA sample and ligating specialised adapters to both fragments ends to make NGS library. Source:https://www.cd-genomics.com/blog/principle-and-workflow-of-illumina-next-generation-sequencing/
Cluster Generation
The prepared library is loaded in a flow cell and placed in the sequencer. A flow cell is a glass slide with 1, 2, or 8 physically separated lanes coated with a lawn of surface-bound, adapter-complimentary oligos. Nowadays, mostly used flow cell is a patterned flow cell produced using semiconductor manufacturing technology. It has a glass substrate containing patterned nano wells with DNA probes that capture the prepared DNA strands for amplification during cluster generation.
 Figure: Illumina Patterened flow cell. Source:https://www.illumina.com/science/technology/next-generation-sequencing/sequencing-technology/patterned-flow-cells.
Figure: Illumina Patterened flow cell. Source:https://www.illumina.com/science/technology/next-generation-sequencing/sequencing-technology/patterned-flow-cells.
Cluster generation begins once the samples are attached to the flow cell and bridge amplification occurs. During this process, the DNA fragments of the library get hybridized with one of the oligonucleotides present on the flow cell surface. This is then followed by generating a complementary strand by elongating the oligo attached to the flow cell by DNA polymerase. The original molecule is washed away, and the strand bends over like a bridge and attaches to the next oligo in the flow cell. This second oligo is complementary to another adapter sequence, and polymerase generates a complementary strand forming a double-stranded bridge. This bridge is denatured, resulting in two single-stranded copies of the molecule tethered to the flow cell. The process is repeated over and over and occurs simultaneously for millions of clusters resulting in clonal amplification of all.
After bridge amplification, the reverse strands are cleaved and washed off, leaving only the forward strands. The 3′ ends are blocked to prevent unwanted priming. When cluster generation is complete, the templates are ready for sequencing.
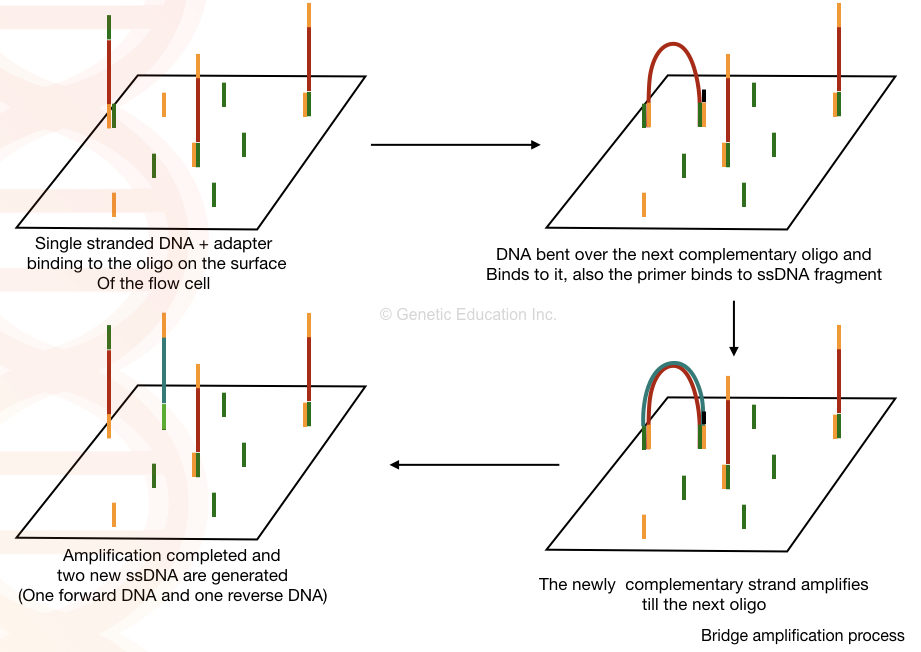 Figure: Bridge Amplification Process. Source: https://geneticeducation.co.in/dna-sequencing-history-steps-methods-applications-and-limitations/
Figure: Bridge Amplification Process. Source: https://geneticeducation.co.in/dna-sequencing-history-steps-methods-applications-and-limitations/
Sequencing by Synthesis
Sequencing coincides for every cluster on a flow cell.The components required for sequencing include sequencing primer, DNA polymerase, and fluorescently labeled with a fluorophore. However, based on the chemistry used in their respective machine (4-channel chemistry, 2-channel chemistry, and 1-channel chemistry), either all the nucleotides are labeled, or only a few nucleotides are labeled with the fluorophore
In 4-channel chemistry, all the nucleotides are labeled with four fluorescent dyes. Two-channel chemistry uses two different fluorescent dyes, while one-channel chemistryuses only one dye.
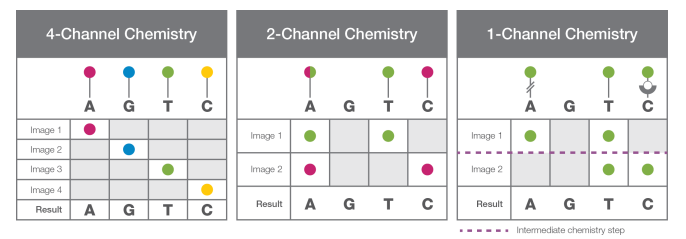 Figure: Four-, Two- and One- Channel Chemistry: Four Channel chemistry uses nucleotides labelled with four different dyes, Two channel chemistry uses two different fluorescent dyes and one channel Chemistry uses only one dye. Source: https://www.illumina.com/content/dam/illumina-marketing/documents/products/techspotlights/cmos-tech-note-770-2013-054.pdf
Figure: Four-, Two- and One- Channel Chemistry: Four Channel chemistry uses nucleotides labelled with four different dyes, Two channel chemistry uses two different fluorescent dyes and one channel Chemistry uses only one dye. Source: https://www.illumina.com/content/dam/illumina-marketing/documents/products/techspotlights/cmos-tech-note-770-2013-054.pdf
In the case of 4-channel chemistry, all the above components are passed through the flow cell, where sequencing primer anneals to its complementary location on the adapter, and DNA polymerase adds the complementary nucleotides labeled with a fluorophore. This fluorophore acts as a blocking group, and DNA polymerase cannot add another nucleotide until it is removed, allowing the detector to record the fluorescence of each base being added. After adding each nucleotide, fluorophore in the cluster are excited by the light source and the characteristic fluorescent signal emitted are recorded. This process is called sequencing-by-synthesis.
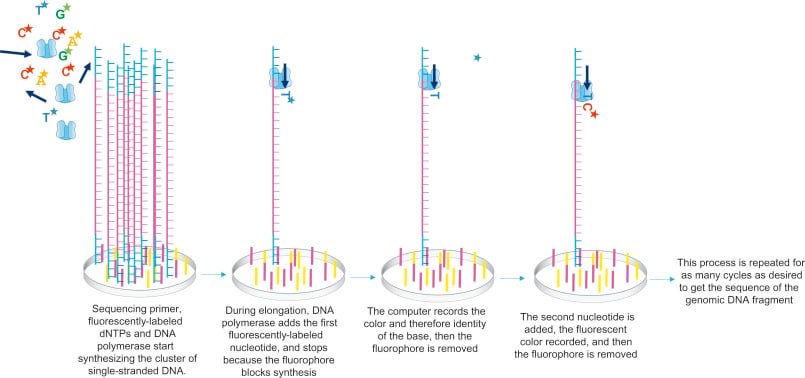 Figure: In the figure, only one strand of DNA is shown from this cluster, while the sequencing occurs simultaneously in the entire cluster. As shown, sequencing primers, fluorescently labelled dNTPs, and DNA polymerase are added. Primer gets attached to the DNA template, which gets elongated by DNA polymerase. Once, the complementary nucleotide is added to the end of the primer, fluorophore blocks DNA polymerase from adding any more nucleotides and fluorophore is recorded by the computer. This fluorophore is then removed and washed away. DNA polymerase then adds the second complementary nucleotide, these cycles are repeated to get a read from this cluster. Source: DNA Sequencing Unit . Molecular Biology.
Figure: In the figure, only one strand of DNA is shown from this cluster, while the sequencing occurs simultaneously in the entire cluster. As shown, sequencing primers, fluorescently labelled dNTPs, and DNA polymerase are added. Primer gets attached to the DNA template, which gets elongated by DNA polymerase. Once, the complementary nucleotide is added to the end of the primer, fluorophore blocks DNA polymerase from adding any more nucleotides and fluorophore is recorded by the computer. This fluorophore is then removed and washed away. DNA polymerase then adds the second complementary nucleotide, these cycles are repeated to get a read from this cluster. Source: DNA Sequencing Unit . Molecular Biology.
How to read the nucleotide in NGS?
During sequencing, different segments of DNA fragments are read at various points, and one cluster contains multiple reads. Once the flow cell is flooded with all the ingredients (DNA polymerase, sequencing primer, and fluorescently labeled nucleotide), the sequencing primer anneals to the left side of genomic DNA, which reads the fragment from the 5’ side and is considered first read or read 1. These ingredients are then washed off, and the 3’ end of the template is deprotected, which allows it to fold over and bind to the oligo on a flow cell.
The flow cell is again flooded with all ingredients, but primers complementary to the correct adapter are added. This decodes the index found in the suitable adapter and is considered a second read or read 2. After each cycle, fluorophore emission is detected for each cluster using an optical system and determines the base incorporated. This process of determining sequence from both ends is called paired-end sequencing. Meanwhile, in single-read sequencing, the sequences are determined from only one end of the library fragment.
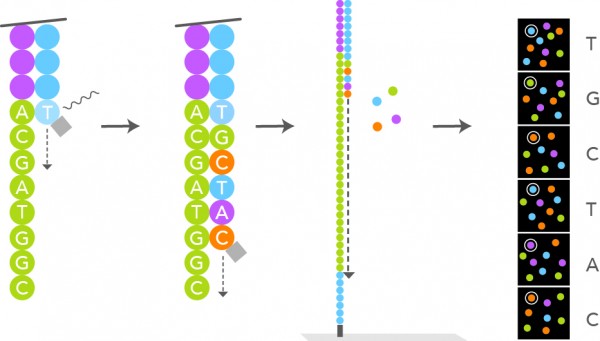 Figure: Addition of fluorescently labelled nucleotide and identifying the fluorophore. Source: https://www.lexogen.com/rna-lexicon-next-generation-sequencing/
Figure: Addition of fluorescently labelled nucleotide and identifying the fluorophore. Source: https://www.lexogen.com/rna-lexicon-next-generation-sequencing/
Data Analysis
After the sequencing is complete, the optical signals are translated to a nucleotide sequence called base calling. The accuracy of base calling is measured by the Phred quality score (Q score), the most common metric for assessing sequencing data quality. Q score indicates the probability that the given base is called incorrectly by the sequencer.
Q scores are defined as property that is logarithmically related to the base calling error probabilities (P)2. Q= -10 log10P
This Q score determines a good base or a bad one. The Quality Score and Base Calling Accuracy is shown below. The scoring Q30 is ideal for a range of sequencing applications.
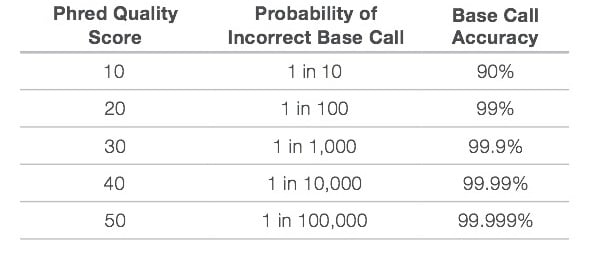 Figure: Quality Score and Base Calling Accuracy. Source:https://www.illumina.com/documents/products/technotes/technote_Q-Scores.pdf
Figure: Quality Score and Base Calling Accuracy. Source:https://www.illumina.com/documents/products/technotes/technote_Q-Scores.pdf
During the library preparation, each sample is given unique index sequences, which are called multiplexing; this allows large numbers of libraries to be pooled together and sequenced simultaneously in a single sequencing run. So, before data analysis, a process called demultiplexing occurs, which separates the sequences from pooled sample libraries based on their unique indexes. For each sample, reads with similar stretches of the bases are locally clustered. Forward and reverse reads are paired, creating contiguous sequences. These newly identified contiguous sequences are aligned to a reference sequence.
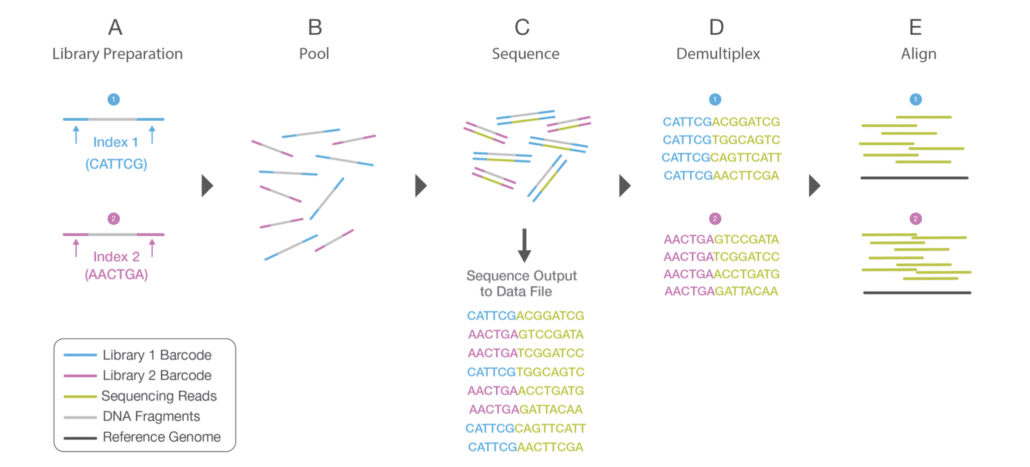 Figure: Multiplexing process is shown in A during Library Preparation, where unique indexes are provided to each sample. After library preparation, each sample is pooled together. The sequencing process is shown in C. After sequencing, the demultiplexing algorithm sorts the reads into different files according to their indexes. Source:https://www.illumina.com/content/dam/illumina-marketing/documents/products/illumina_sequencing_introduction.pdf
Figure: Multiplexing process is shown in A during Library Preparation, where unique indexes are provided to each sample. After library preparation, each sample is pooled together. The sequencing process is shown in C. After sequencing, the demultiplexing algorithm sorts the reads into different files according to their indexes. Source:https://www.illumina.com/content/dam/illumina-marketing/documents/products/illumina_sequencing_introduction.pdf
The number of reads that align with the reference genome is vital, called coverage depth. The higher the coverage depth more number have the same sequence and is aligned. This alignment shows differences or similarities between the reference genome and the sequence of isolated samples that can be identified using bioinformatics tools.
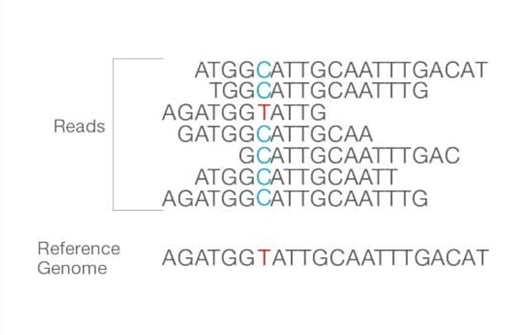 Figure: Reads are aligned to the reference sequence. After the alignment, the differences between them can be identified. Source: Illumina sites
Figure: Reads are aligned to the reference sequence. After the alignment, the differences between them can be identified. Source: Illumina sites
Following alignment, many analysis variations are possible, such as single nucleotide polymorphism (SNP) or insertion-deletion (indel) identification, read counting for RNA methods, phylogenetic or metagenomic analysis, and more.
Application of Next-Generation Sequencing
Next Generation Sequencing technology has a variety of applications. Some of these are mentioned below:
- NGS can be used to perform metagenomic sequencing (metagenomic sequencing is a process of identifying organisms from environmental or clinical samples by using multiple sets of primers for different organisms) for the detection of unknown disease-associated viruses and novel human viruses.
- In humans, less than 2% of the genome consists of an exome that contains most of the known disease-causing variants, and with NGS whole exome sequencing is cost-effective.
- This method is used to sequence not only human genomes but also other species such as agriculturally important livestock, plants, or disease-related microbes.
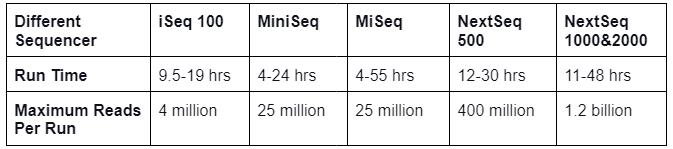
- Whole genome sequencing analyses the entire genome and produces large volumes of data making it a powerful tool for genomics research. Different sequencers are present which sequence the sample in a short period of time and are mentioned below:
 ## Limitation of Next-Generation Sequencing
## Limitation of Next-Generation Sequencing
There are a few limitations of NGS, which are mentioned below:
- It can be costly as it requires sophisticated bioinformatics systems, fast data processing, and large data storage capabilities.
- PCR amplification prior to sequencing, may lead to PCR biases during library preparation (sequence GC-content, fragment length, and false diversity) and analysis (base errors/favoring certain sequences over others).
References
- Clark D, Pazdernik N, McGehee M. DNA Sequencing Unit. Molecular Biology. 2019. 240–269 p.
- https://www.illumina.com/science/technology/next-generation-sequencing/beginners/ngs-workflow.html
- https://medium.com/@tiffanysouterre/dna-sequencing-techniques-explained-53c21eef51b1
- https://www.cd-genomics.com/blog/principle-and-workflow-of-illumina-next-generation-sequencing/
- Illumina sites
- https://www.technologynetworks.com/genomics/articles/an-overview-of-next-generation-sequencing

Tankeshwar Acharya, MSc (Medical Microbiology)
Tankeshwar Acharya is an Assistant Professor in the Department of Microbiology at Patan Academy of Health Sciences (PAHS), Nepal, where he has been teaching and practicing clinical microbiology for over 14 years. He is the founder of Microbe Online, one of the leading free microbiology education resources on the web, covering bacteriology, mycology, parasitology, immunology, and clinical laboratory diagnostics written from direct experience in both the classroom and the diagnostic laboratory.