HIV: Structure, Laboratory Diagnosis, and Natural Resistance
Complete guide to HIV — structure (gp120, gp41, p24, reverse transcriptase), laboratory diagnosis (ELISA, Western blot, PCR, CD4 count), and why some people are naturally resistant to HIV infection (CCR5-delta32 mutation).
In 1981, clinicians in Los Angeles noticed something that shouldn't have been possible: young, otherwise healthy men developing Pneumocystis jirovecii pneumonia (then still called P. carinii pneumonia; the species was renamed in 2002), a fungal infection that had previously only been seen in severely immunocompromised patients; people on chemotherapy or organ transplant recipients. The immune systems of these men weren't just failing, they were failing in a way that looked like the CD4+ T lymphocytes, the cells that coordinate virtually the entire adaptive immune response, had been systematically eliminated.
The reason turned out to be a virus that had evolved a strategy unlike almost anything seen before: rather than simply killing host cells through brute replication, HIV infects and gradually depletes the very cells the immune system depends on to recognize and respond to infection. Lose enough CD4+ T cells, and the body cannot mount effective responses to pathogens it normally handles effortlessly; fungi, parasites, and viruses that would never cause disease in a healthy person become life-threatening. This is acquired immunodeficiency syndrome (AIDS), not a disease caused directly by the virus, but a collapse of immune surveillance caused by the virus's slow destruction of its own host cell target over years.
Understanding HIV from a clinical microbiology standpoint requires three things: knowing how the virus is built (which predicts which drug classes can target it), knowing how infection is diagnosed in the laboratory (which requires understanding why standard antibody tests fail in early infection and in infants), and understanding why some individuals are genetically protected from infection at all. This article covers all three.
Structure of HIV
HIV belongs to the genus Lentivirus within the family Retroviridae. Two types exist; HIV-1 (the predominant global cause of AIDS) and HIV-2 (less virulent, mainly found in West Africa). The virus is approximately 100–120 nm in diameter with a characteristic dense, cone-shaped nucleocapsid.
Overall structure
HIV has the following structural layers from outside to inside:
1. Lipid bilayer envelope The outermost layer is derived from the host cell membrane during the budding process. It contains both viral glycoproteins and host cell proteins acquired during budding.
2. Envelope glycoproteins (Env); gp120 and gp41 72 external spikes are embedded in the envelope, each composed of two major glycoproteins:
- gp120 (surface unit) — binds to the CD4 receptor on CD4+ T lymphocytes and cells of the monocyte/macrophage lineage. Also binds to chemokine coreceptors CCR5 and CXCR4. HIV-1 and HIV-2 are distinguished serologically by differences in gp120. On the basis of gp120 gene variability (V3 region), HIV-1 is further classified into subtypes (clades A through I)
- gp41 (transmembrane unit) — mediates fusion between the viral membrane and the host cell membrane, allowing viral entry
3. Matrix protein (p17) Beneath the viral envelope, p17 maintains the structural integrity of the virion and plays a role in nuclear transport of the pre-integration complex.
4. Capsid (p24) A protein shell enclosing the viral RNA and associated enzymes. The p24 antigen is detectable in blood during acute infection before antibodies develop — the basis of the p24 antigen test in early HIV diagnosis.
5. Nucleocapsid proteins (p7, p9) Enclose the viral RNA genome within the capsid during assembly.
6. Viral RNA genome Two identical copies of a 9.8 kb single-stranded positive-polarity RNA genome. HIV is a retrovirus — its RNA genome must be reverse transcribed into DNA before integration into the host genome.
Viral enzymes
Three enzymes are packaged inside the HIV virion, all encoded by the pol gene and all targets of antiretroviral therapy:
| Enzyme | Function | Drug class targeting it |
|---|---|---|
| Reverse transcriptase (RT) | Converts viral RNA genome into double-stranded DNA | NRTIs, NNRTIs |
| Integrase (IN) | Integrates viral DNA into host cell chromosome | Integrase inhibitors (raltegravir, dolutegravir) |
| Protease (PR) | Cleaves viral polyproteins into functional proteins during virion maturation | Protease inhibitors (ritonavir, lopinavir) |
Structural and regulatory genes
HIV encodes 3 structural genes and 6 regulatory genes:
Structural genes:
- Gag — encodes core proteins: p24 (capsid), p17 (matrix), p7/p9 (nucleocapsid)
- Env — encodes envelope glycoproteins: gp120 and gp41
- Pol — encodes viral enzymes: reverse transcriptase, integrase, protease
Regulatory genes (required for replication):
- Tat — activates transcription of viral genes
- Rev — transports late mRNAs from nucleus to cytoplasm
Regulatory genes (not required for replication):
- Nef — decreases CD4 and MHC class I expression on infected cells. Deletions/mutations in the nef gene are found in some long-term non-progressors — individuals infected with HIV who do not progress to AIDS
- Vif — enhances viral infectivity
- Vpr — transports viral core from cytoplasm into nucleus
- Vpu — enhances virion release from the cell
Pathogenesis of HIV Infection and AIDS
Understanding why HIV causes AIDS, rather than just acute illness, requires following what happens after the virus first enters the body — not just structurally, but in terms of the immune system's long, losing battle to contain it.
Entry and acute infection
HIV is transmitted primarily through sexual contact, exposure to infected blood, or vertically from mother to child during delivery or breastfeeding. The transmitted virus is almost always R5-tropic — using the CCR5 coreceptor (see Part 4 for why this matters for natural resistance). Entry follows the two-step receptor binding sequence described in the Structure section: gp120 binds CD4, conformational change exposes the coreceptor binding site, gp120 then binds CCR5 or CXCR4, and gp41 mediates membrane fusion.
Once inside a CD4+ T lymphocyte or macrophage, reverse transcriptase converts the viral RNA genome into double-stranded DNA, which integrase then inserts into the host cell chromosome as a provirus. This integrated provirus can remain transcriptionally silent for years (latency) or actively transcribe new viral RNA and proteins, eventually producing progeny virions that bud out to infect new cells.
2–4 weeks after infection, most people experience an acute retroviral syndrome: fever, lymphadenopathy, sore throat, rash, myalgia, and malaise, resembling infectious mononucleosis. During this phase, viral load in blood is extremely high, CD4+ counts drop sharply, and the person is highly infectious. The immune system mounts a vigorous response, particularly through cytotoxic CD8+ T cells, which partially controls viral replication. Viral load falls, CD4+ counts partially recover, and the person enters the asymptomatic chronic phase. Many people don't recognise or seek care for this acute phase, which is why transmission frequently occurs here without knowledge.
Chronic infection and CD4 depletion: the slow war of attrition
During the chronic asymptomatic phase, which lasts an average of 8–10 years without treatment, HIV continues to replicate at a high rate (roughly 10 billion new virions produced and cleared daily), and CD4+ T cells continue to be destroyed at a rate that gradually exceeds the body's ability to replace them. The mechanisms of CD4+ T cell depletion are multiple:
- Direct viral cytopathic effect: virus budding disrupts the cell membrane; accumulated viral proteins are toxic to the cell; infected cells are killed by the immune response (CD8+ cytotoxic T cells recognising infected cells)
- Bystander apoptosis: uninfected CD4+ T cells undergo apoptosis after contact with gp120 or with viral proteins shed from infected cells
- Immune activation and exhaustion: the chronic immune response to persistent infection accelerates CD4+ T cell turnover and eventual exhaustion
The result is a steady decline in CD4+ count — normally 500–1500 cells/μL — falling at roughly 50–100 cells/μL per year on average. The CD4 count becomes the primary clinical marker of how far this depletion has progressed.
AIDS: the consequence of immune collapse
When the CD4+ count falls below 200 cells/μL, the adaptive immune response is sufficiently compromised that pathogens normally controlled effortlessly begin to cause life-threatening disease. This threshold defines AIDS (acquired immunodeficiency syndrome) clinically. The characteristic illnesses are AIDS-defining opportunistic infections — infections that essentially never occur in immunocompetent individuals:
- CD4 < 200 cells/μL: Pneumocystis jirovecii pneumonia (PCP), toxoplasmosis, cryptosporidiosis, disseminated histoplasmosis
- CD4 < 100 cells/μL: Cryptococcal meningitis, CMV retinitis
- CD4 < 50 cells/μL: Disseminated Mycobacterium avium complex (MAC), CMV disease, CNS lymphoma
HIV itself does not directly cause these pneumonias, meningitides, or lymphomas; it causes them indirectly by dismantling the immune surveillance that would normally prevent them. This is why HIV is unlike most pathogens: its primary clinical damage is not what the virus does to tissues directly, but what the immune system can no longer do once the virus has done its work on CD4+ T cells.
Why antiretroviral therapy (ART) works
ART does not eliminate the integrated provirus from infected cells; it suppresses active viral replication by blocking one or more of the three viral enzymes (reverse transcriptase, integrase, protease) described in Part 1. With viral replication suppressed, CD4+ T cell destruction slows, counts stabilise or recover, and the immune system retains enough function to control opportunistic pathogens. This is why patients on effective ART can live near-normal lifespans despite remaining HIV-positive, the provirus persists, but active replication and CD4 depletion are held in check.
Laboratory Diagnosis of HIV Infection
Laboratory tests for HIV diagnosis are divided into four categories: antigen/antibody detection, virus cultivation, antibody confirmation, and viral genome amplification.
Sample collection and transport
- Standard HIV-1/HIV-2 antibody testing requires a single tube (10 mL) of whole blood
- Specimens stored at room temperature: stable for up to 3 days
- Specimens stored at 4°C: stable for up to 7 days
- For longer storage, serum or plasma must be separated and stored at −20°C
- Specimens for PCR must be processed within 48 hours — viral RNA/DNA degrades over time
Window period
Antibodies to HIV become detectable 3 to 12 weeks after infection. This gap between infection and antibody detectability is the window period — a critical time when a recently infected person may test antibody-negative despite carrying the virus. Modern 4th-generation combination antigen/antibody tests have reduced the window period to less than 3 weeks by detecting both p24 antigen (present from day 14–21) and HIV antibodies simultaneously.
Diagnostic algorithm
Step 1 — Screening test: 4th generation ELISA (combination Ag/Ab assay) The current standard for HIV screening. Detects both HIV-1/HIV-2 antibodies and HIV-1 p24 antigen simultaneously. Highly sensitive (>99.5%) but false positives occur. Any reactive result must be confirmed.
Step 2 — Confirmatory test: Western blot or HIV-1/HIV-2 differentiation immunoassay A positive Western blot requires bands at specific HIV protein positions — at minimum, two bands from: p24, gp41, gp120/160. An indeterminate Western blot requires further follow-up testing. Modern HIV differentiation immunoassays (identifying HIV-1 vs HIV-2) are now preferred over Western blot in many reference laboratories.
Step 3 — Nucleic acid test (NAT/PCR) for indeterminate or discordant results
Individual diagnostic tests in detail
Point of care (rapid) tests Immunochromatographic tests that can be performed in 15–30 minutes at the bedside or in resource-limited settings. Detect HIV-1/HIV-2 antibodies. High sensitivity and specificity but all positives must be confirmed with laboratory testing. Widely used in antenatal screening programs and voluntary counseling and testing centers.
ELISA (Enzyme-Linked Immunosorbent Assay) The most widely used HIV screening test. 4th generation assays detect both p24 antigen and IgM/IgG antibodies. False positives can occur in autoimmune conditions, pregnancy, malaria, and recent influenza vaccination. All repeatedly reactive ELISA results must be confirmed by Western blot or NAT.
p24 Antigen testing p24 antigen appears in blood 14–21 days after infection — before antibodies develop. Particularly useful:
- For patients who are high risk and symptomatic but HIV antibody-negative (early acute infection during window period)
- For specimens that are ELISA-positive but Western blot-negative or indeterminate
- For confirming HIV infection in neonates born to HIV-positive mothers — maternal IgG antibodies cross the placenta and make antibody-based testing unreliable in infants under 18 months
Western blot Highly specific confirmatory test. Viral proteins are separated by electrophoresis, transferred to a membrane, and probed with patient serum. Specific antibody bands at positions corresponding to HIV structural proteins (gp160, gp120, gp41, p66, p51, p31, p24, p17) indicate HIV infection. A positive result requires reactivity at a minimum of two envelope bands.
HIV PCR (Nucleic Acid Amplification Test) Detects HIV RNA (viral load test) or HIV proviral DNA in blood. Uses reverse transcription PCR (RT-PCR). Key applications:
- Diagnosis in infants — PCR is the standard for diagnosing HIV in infants born to HIV-positive mothers, tested at 14–21 days, 1–2 months, and 4–6 months of age
- Resolving indeterminate Western blot results
- Testing immunocompromised patients who may not mount an antibody response
- Viral load monitoring — quantitative HIV RNA PCR monitors disease progression and treatment response in HIV-positive patients
Virus cultivation HIV can be isolated from peripheral blood mononuclear cells (PBMCs) co-cultivated with activated PBMCs from HIV-negative donors in the presence of IL-2. A positive result is detected by p24 antigen or reverse transcriptase activity in the culture medium. This is not performed in routine diagnostic laboratories — used only in research settings.
CD4+ lymphocyte count
A hallmark of chronic HIV infection is the progressive depletion of CD4+ T lymphocytes. CD4 count monitoring is essential for:
- Clinical staging of HIV infection
- Deciding when to initiate antiretroviral therapy
- Initiating prophylaxis against opportunistic infections (e.g. Pneumocystis jirovecii pneumonia prophylaxis when CD4 < 200 cells/μL)
- Monitoring treatment response
| CD4 count | Clinical significance |
|---|---|
| > 500 cells/μL | Normal or early infection; no opportunistic infections |
| 200–500 cells/μL | Moderate immunosuppression; increased risk |
| < 200 cells/μL | Severe immunosuppression — AIDS-defining threshold; PCP prophylaxis indicated |
| < 50 cells/μL | Profound immunosuppression; high risk of CMV, MAC, CNS lymphoma |
Natural Resistance to HIV Infection
Not everyone exposed to HIV becomes infected. Some individuals — particularly those repeatedly exposed through high-risk behavior — remain HIV-negative. While safe sex practices and needle safety explain many cases, a subset of people appear to have genetic protection against HIV infection.
How HIV enters a cell — the receptor problem
For a virus to infect a cell, it must first attach firmly to it. HIV requires two sequential binding steps to enter a CD4+ T lymphocyte:
- Primary receptor binding: gp120 on the HIV surface binds to the CD4 receptor on the host cell. This causes a conformational change in gp120 that exposes the coreceptor binding site
- Coreceptor binding: The exposed gp120 then binds to a chemokine coreceptor — either CCR5 or CXCR4 — on the same cell surface
- Membrane fusion: gp41 mediates fusion between viral and cellular membranes, allowing viral entry
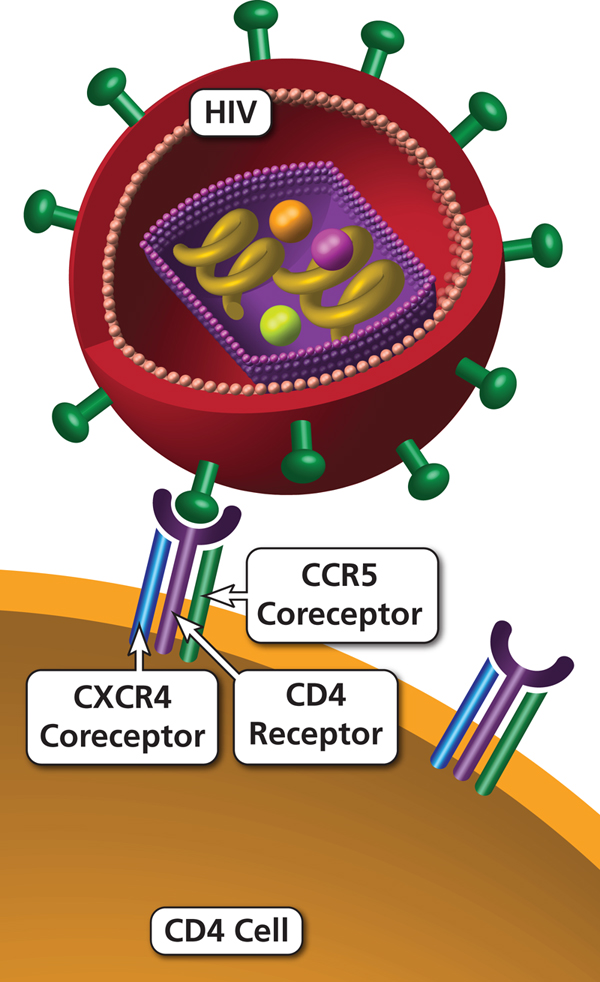 If either the CD4 receptor or the required coreceptor is absent from the cell surface, HIV cannot enter — the entire infection process stops at step one.
If either the CD4 receptor or the required coreceptor is absent from the cell surface, HIV cannot enter — the entire infection process stops at step one.
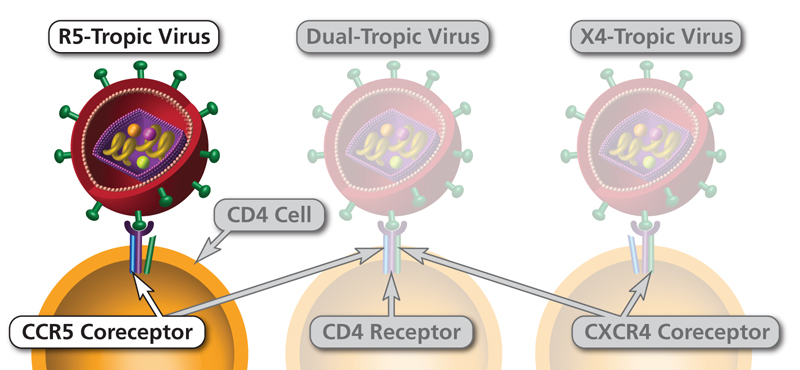
HIV uses different coreceptors at different stages:
- Early infection: HIV is predominantly R5-tropic — uses CCR5 coreceptor. Transmitted via sexual contact, blood, or vertical transmission
- Late infection/AIDS: HIV may switch to X4-tropic (CXCR4) or dual-tropic (both), correlating with more rapid CD4 decline
The CCR5-Δ32 mutation — genetic resistance to HIV
Studies have found that a specific mutation in the gene encoding CCR5 — called CCR5-Δ32 (delta 32, a 32-base pair deletion) — produces a truncated, non-functional CCR5 protein that is not expressed on the cell surface. Without surface CCR5, R5-tropic HIV (which accounts for almost all sexually transmitted HIV) cannot enter CD4+ T cells.
The clinical significance:
- Homozygotes (two copies of CCR5-Δ32): Essentially completely resistant to R5-tropic HIV infection. Even with repeated high-risk exposure, these individuals do not become infected with the predominant strains of HIV
- Heterozygotes (one copy of CCR5-Δ32): Not fully protected from infection, but if infected, disease progresses significantly more slowly — lower viral loads, slower CD4 decline, longer time to AIDS
This mutation is found predominantly in people of Western European ancestry:
- ~1% are homozygous (fully resistant)
- ~10–15% are heterozygous (partial protection)
The mutation is rare in East Asian, African, and South Asian populations, which may partly explain regional differences in HIV transmission dynamics.
Clinical application — CCR5 inhibitors
The discovery of CCR5 as an essential HIV coreceptor led directly to the development of CCR5 inhibitor antiretroviral drugs — a class of drugs that block the CCR5 receptor on CD4+ cells, preventing R5-tropic HIV from attaching. Maraviroc is the first approved CCR5 inhibitor (2007), used in patients with confirmed R5-tropic HIV infection.
The Berlin Patient — first cure of HIV
Timothy Ray Brown, known as "The Berlin Patient," is recognized as the first person functionally cured of HIV.
Brown was diagnosed with HIV in 1995. In 2006, he was also diagnosed with acute myeloid leukemia. His German physicians made a remarkable treatment decision; they selected a bone marrow donor who was homozygous for CCR5-Δ32.
After two stem cell transplants from this CCR5-Δ32 homozygous donor:
- His leukemia was cured
- His immune system was reconstituted with donor cells lacking functional CCR5
- HIV was undetectable in his blood and tissues — and remained so until his death from leukemia relapse in 2020
Brown's case proved that eliminating CCR5 expression from immune cells can effectively cure HIV infection, and it remains the foundational case study for ongoing gene therapy and stem cell approaches to HIV cure research.
Brown later founded the Timothy Ray Brown Foundation in Washington, DC, dedicated to HIV/AIDS cure research.
How to Remember
Three enzymes, three drug classes, one gene that encodes all three. The pol gene encodes reverse transcriptase, integrase, and protease — and each has its own drug class targeting it (NRTIs/NNRTIs for RT, integrase inhibitors, protease inhibitors). A useful anchor: pol is the "pharmacology gene" because every drug class in antiretroviral therapy maps back to one of its three products. If an exam question describes the mechanism of a named antiretroviral drug, asking "which enzyme does this block?" and matching to one of these three is the fastest path to the right answer.
gp120 = attachment, gp41 = fusion, p24 = the shell you can test for. The three structural proteins students most need to keep separate: gp120 grabs the CD4 receptor (attachment step), gp41 pulls the viral membrane against the cell membrane and drives them together (fusion step), and p24 is the capsid protein encasing the viral RNA — detectable in blood before antibodies appear, making it the early diagnostic antigen. If you remember "120 attaches, 41 fuses, 24 you can find first in the blood," the three most-tested HIV surface/structural proteins stop blurring together.
Window period = the gap before the immune photograph develops. The window period exists because antibody production requires weeks of clonal expansion and class switching after initial antigen exposure. Think of it as a photograph being developed in a darkroom: the image (the infection) was captured the moment of exposure, but the photograph (the antibody) doesn't appear until the development process completes. During that development period, a standard antibody-only test shows nothing. The 4th-generation test shortens this gap by also detecting p24 antigen — the viral protein that appears two weeks earlier, before any antibody is made.
CCR5 is the door HIV needs to open early; CXCR4 is the door it learns to use later. Early (transmitted) HIV is almost always R5-tropic because CCR5 is expressed on macrophages and memory T cells at mucosal surfaces — the first cells HIV encounters after sexual transmission. Later in infection, the virus can evolve to use CXCR4 (X4-tropic), which is expressed on naïve T cells; this switch correlates with faster CD4 decline and more rapid progression to AIDS. Knowing this sequence explains why CCR5 inhibitors like maraviroc are most useful in R5-tropic infection (confirmed by tropism testing) and why they lose effectiveness if the virus has already switched.
Key Exam Facts Table
| Feature | Detail |
|---|---|
| Family / Genus | Retroviridae / Lentivirus |
| Types | HIV-1 (global, more virulent); HIV-2 (mainly West Africa, less virulent) |
| Genome | 2 identical copies of ssRNA (+), 9.8 kb; requires reverse transcription to DNA for integration |
| Size | ~100–120 nm |
| Envelope | Present; 72 gp120/gp41 spikes |
| Primary receptor | CD4 (on CD4+ T lymphocytes and monocyte/macrophage lineage) |
| Coreceptors | CCR5 (early/R5-tropic HIV); CXCR4 (late/X4-tropic HIV) |
| Key enzymatic drug targets | Reverse transcriptase (NRTIs, NNRTIs); Integrase (raltegravir, dolutegravir); Protease (ritonavir, lopinavir) |
| Incubation to AIDS | Average 8–10 years without treatment |
| Acute retroviral syndrome | 2–4 weeks post-infection; fever, lymphadenopathy, rash (mononucleosis-like) |
| AIDS-defining CD4 threshold | < 200 cells/μL |
| Window period | 3–12 weeks (antibody only); reduced to < 3 weeks with 4th-generation Ag/Ab test |
| Earliest detectable marker | p24 antigen (14–21 days post-infection) |
| Screening test | 4th-generation ELISA (combined p24 Ag + HIV-1/HIV-2 Ab) |
| Confirmatory test | Western blot (requires ≥2 bands: p24, gp41, gp120/160) or HIV-1/HIV-2 differentiation immunoassay |
| Infant diagnosis | PCR (not antibody tests — maternal IgG crosses placenta and persists up to 18 months) |
| Natural resistance mechanism | CCR5-Δ32 homozygosity: truncated non-functional CCR5, R5-tropic HIV cannot enter CD4+ T cells |
| CCR5-Δ32 population frequency | ~1% homozygous (fully resistant); ~10–15% heterozygous (slower progression) in Western European ancestry |
| CCR5 inhibitor drug | Maraviroc (first approved 2007, for confirmed R5-tropic HIV) |
Where Students Get Confused
"A negative HIV test means the person isn't infected." Not during the window period. A standard antibody-only test can be completely negative in a person who was infected 2–4 weeks ago and is currently at peak viremia during acute retroviral syndrome, the most infectious phase of the disease. A 4th-generation test reduces but doesn't eliminate this gap (p24 becomes detectable by day 14–21). Any high-risk exposure within the past 12 weeks warrants repeat testing regardless of an initial negative result.
"HIV-2 behaves identically to HIV-1 in testing and disease." It doesn't on either count. HIV-2 is less virulent, progresses to AIDS more slowly, and has naturally lower viral loads. More importantly for diagnosis: some rapid antibody tests and older ELISAs may not reliably detect HIV-2 antibodies. Current 4th-generation assays are designed to detect both, but this is worth knowing, particularly in a clinical setting seeing patients from West Africa where HIV-2 prevalence is higher.
"Western blot is the best confirmation test." It was the standard for decades, but modern HIV-1/HIV-2 differentiation immunoassays are now preferred over Western blot in many reference laboratories, because they also distinguish HIV-1 from HIV-2, which Western blot cannot reliably do. An indeterminate Western blot result is not rare, and doesn't mean the patient is negative — it requires follow-up with NAT.
"CD4 count tells you if someone is infected with HIV." It doesn't. CD4 count is not a diagnostic test; it measures the degree of immunosuppression in someone already known to be HIV-positive. A low CD4 count has many causes beyond HIV. PCR and serology are the diagnostic tools; CD4 count is the staging and monitoring tool.
"ART cures HIV." It doesn't. Antiretroviral therapy suppresses viral replication to undetectable levels, which prevents CD4 depletion and AIDS-defining illness, but the integrated provirus persists in resting CD4+ T cells (the latent reservoir) indefinitely. Stopping ART leads to viral rebound within weeks. Functional cure (like the Berlin Patient) requires eliminating the reservoir, which remains an active research challenge.
References and Further Reading
- Tille, P. M. (2017). Bailey & Scott's Diagnostic Microbiology (14th ed.). Mosby Elsevier.
- Turner, B. G., & Summers, M. F. (1999). Structural biology of HIV. Journal of Molecular Biology, 285(1), 1–32. https://doi.org/10.1006/jmbi.1998.2354
- Buttò, S., Suligoi, B., Fanales-Belasio, E., & Raimondo, M. (2010). Laboratory diagnostics for HIV infection. Annali dell'Istituto superiore di sanità, 46(1), 24–33. https://doi.org/10.4415/ANN_10_01_04
- Fearon, M. (2005). The laboratory diagnosis of HIV infections. Canadian Journal of Infectious Diseases and Medical Microbiology, 16(1), 26–30. https://doi.org/10.1155/2005/515063
- Samson, M., Libert, F., Doranz, B. J., et al. (1996). Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature, 382(6593), 722–725. https://doi.org/10.1038/382722a0
- Biasin, M., De Luca, M., Gnudi, F., & Clerici, M. (2013). The genetic basis of resistance to HIV infection and disease progression. Expert Review of Clinical Immunology, 9(4), 319–334. https://doi.org/10.1586/eci.13.16
- WHO. (2019). Consolidated Guidelines on HIV Testing Services. World Health Organization.
- Deeks, S. G., Overbaugh, J., Phillips, A., & Buchbinder, S. (2015). HIV infection. Nature Reviews Disease Primers, 1, 15035. https://doi.org/10.1038/nrdp.2015.35
Frequently Asked Questions
What is the difference between HIV-1 and HIV-2?
What is the HIV testing window period?
Why can't antibody tests diagnose HIV in newborns?
What does the CCR5-delta32 mutation do?
What is the difference between viral load and CD4 count?
What is the role of the Nef protein?
How does HIV cause AIDS if the virus itself doesn't directly destroy most organs?
Why does it take years for HIV infection to progress to AIDS?
Why can't HIV be cured by stopping antiretroviral therapy once viral load is undetectable?
Why are babies born to HIV-positive mothers tested differently than adults?

Tankeshwar Acharya, MSc (Medical Microbiology)
Tankeshwar Acharya is an Assistant Professor in the Department of Microbiology at Patan Academy of Health Sciences (PAHS), Nepal, where he has been teaching and practicing clinical microbiology for over 14 years. He is the founder of Microbe Online, one of the leading free microbiology education resources on the web, covering bacteriology, mycology, parasitology, immunology, and clinical laboratory diagnostics written from direct experience in both the classroom and the diagnostic laboratory.