Pulsed-Field Gel Electrophoresis (PFGE): Steps, Applications
Pulsed-field gel electrophoresis (PFGE) separates DNA fragments up to 10 Mb by switching the electric field between directions, forcing large molecules to reorient. Learn why pulsing works, the plug-based steps, and why PFGE was the gold standard for outbreak fingerprinting.
In 2011, people across Germany began falling ill with bloody diarrhea, and then with something far worse: hundreds developed hemolytic uremic syndrome, their kidneys failing as their red cells were shredded. More than fifty people would die. The culprit was a Shiga-toxin-producing E. coli, but that was not the useful question. Thousands of people eat E. coli every day. The useful question was: is the strain from this patient in Hamburg the same strain as the one from that patient in Frankfurt, and if so, what did they both eat?
You cannot answer that with a Gram stain, or a biochemical panel, or an antibiogram. Two E. coli that look identical under every routine test can be genetically unrelated, and two isolates of the same outbreak strain can differ slightly in their resistance profile. To link cases, you need to compare the organisms at the level of their genomes.
Here is the problem. An E. coli chromosome is about 5 million base pairs of DNA, wound into a single enormous circle. Drop it onto an ordinary agarose gel and nothing useful happens. Above about 50,000 base pairs, every fragment, whatever its size, snakes through the gel pores at the same rate and piles into a single smear at the top. A conventional gel is blind to the difference between a 100 kb fragment and a 2 Mb one.
So the laboratory does something clever. It embeds the whole bacteria in little blocks of agarose, lyses them gently in place so the giant chromosome is never sheared, and cuts that chromosome with an enzyme chosen precisely because it cuts rarely, only where a specific 6-to-8-base sequence occurs. The 5 Mb chromosome falls into perhaps fifteen or twenty huge pieces.
Now those pieces must be separated by size. And this is where an ordinary gel gives up, because all of them are far too large to sieve.
The trick is to stop pushing the DNA in one direction and start making it change its mind. The field is switched: first at 60 degrees to one side, then 60 degrees to the other, over and over. Each time the field switches, every DNA molecule has to stop, reorient itself, and set off in the new direction. And here is the key: a small molecule turns around quickly; a large one turns around slowly. In the time a large fragment is still laboriously reorienting, a small one has already turned and made progress. Repeat this thousands of times over two or three days, and the fragments spread out along the gel in order of size, a ladder of huge pieces that a conventional gel could never resolve.
The result is a pattern of ten or twenty bands: a DNA fingerprint of the whole genome. The Hamburg isolate and the Frankfurt isolate are run side by side. If the barcodes match, the two patients were infected by the same strain, and the epidemiologists start looking for what they ate in common. In 2011 the answer, eventually, was contaminated fenugreek sprouts.
That is PFGE. It answers a question no phenotypic test can: are these two organisms the same? For thirty years it was the tool that turned a scatter of sick people into a traceable outbreak.
What is pulsed-field gel electrophoresis?
Pulsed-field gel electrophoresis (PFGE) is a technique for separating very large DNA molecules, up to about 10 megabases, by periodically switching the direction of the electric field applied across an agarose gel. **Why the pulsing is necessary: in a conventional gel, DNA fragments larger than about 50 kb all migrate at the same rate, regardless of size, so they cannot be separated. PFGE overcomes this by repeatedly forcing the DNA to change direction. Each time the field switches, a molecule must reorient itself before it can move on, and larger molecules take longer to reorient than smaller ones. Over a run of two to three days, this size-dependent delay spreads the fragments out along the gel, producing a banding pattern that acts as a genome-wide fingerprint.
Pulsed-field gel electrophoresis (PFGE) is a powerful molecular typing technique by which genomic DNA is isolated from the organism of interest, followed by restriction enzyme analysis. The digestion products are then analyzed on an agarose gel by applying an electric field that periodically changes direction allowing for separation of the larger DNA fragments (entire genomic DNA) and approximate measurement of fragment length.
PFGE can separate large DNA molecules (up to 10 Mb), whereas standard DNA gel electrophoresis commonly resolves fragments up to ∼50 kb. PFGE takes 2–3 days, excluding sample preparation.
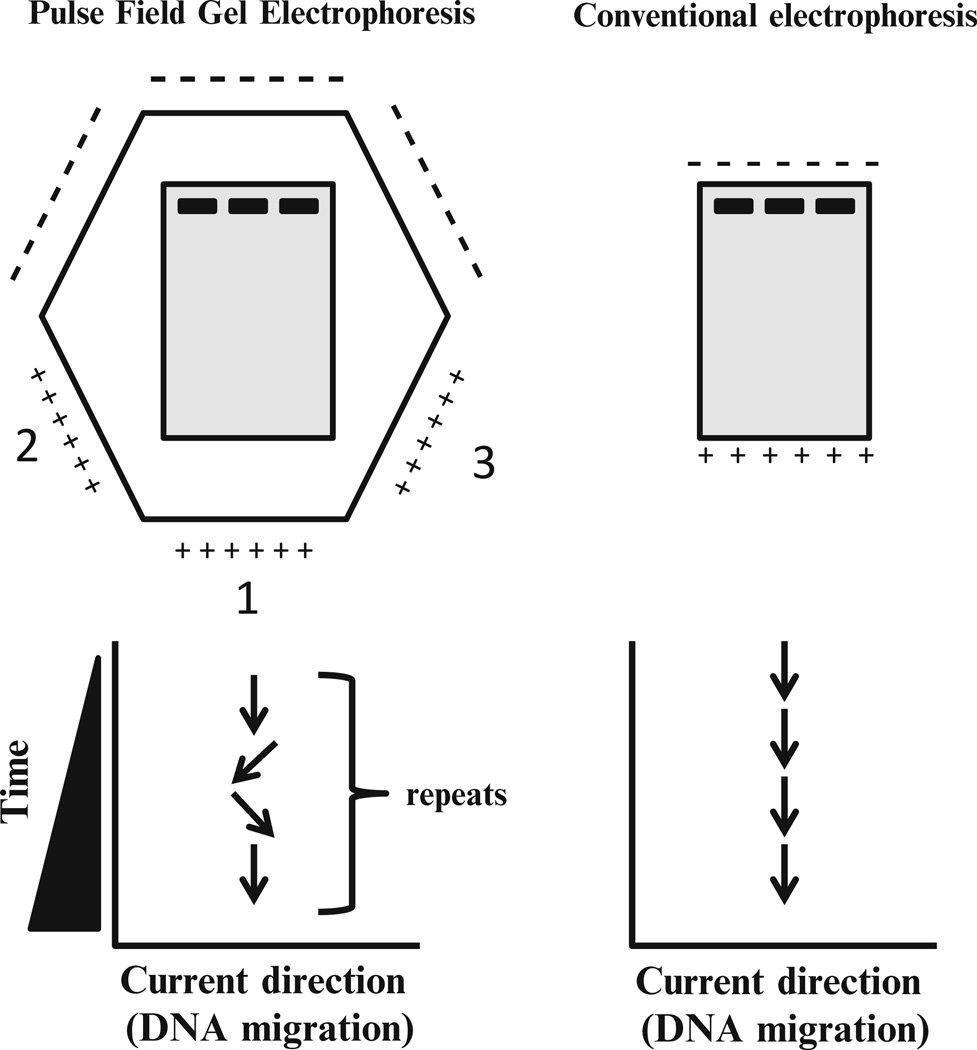 Figure: PFGE vs Conventional electrophoresis
Figure: PFGE vs Conventional electrophoresis
PFGE was first developed by Schwartz and Cantor in 1984 to separate intact yeast chromosomes, which were far too large for conventional gels. The largest molecule resolved by pulsed-field gel electrophoresis has been estimated at around 14 Mb (Barry and Pollard, 1993).
PFGE is a variation of agarose gel electrophoresis. Some of the major differences between conventional gel electrophoresis and PFGE is tabulated here:
Property | Conventional gel electrophoresis | Pulsed-field gel electrophoresis |
Resolving power (Separating | Separates DNA fragments up to ∼50 kb in size. | can separate large DNA molecules(up to 10 Mb) |
Direction of current | The field runs in a single, constant direction throughout the run. | The field direction is periodically switched, alternating between two orientations set about 120° apart, forcing large DNA molecules to reorient at each switch. |
Principle of PFGE
Why a conventional gel cannot separate large DNA
In ordinary agarose gel electrophoresis, small DNA fragments are sieved: a small fragment threads through the gel pores easily and travels fast, a larger one is held back, and so they separate by size. This works well up to roughly 50 kb.
Above that size, the mechanism breaks down. A DNA molecule much larger than the gel pores can no longer pass through as a compact coil. Instead it stretches out and moves end-first, snaking through the pores in a reptile-like motion called reptation. Crucially, once a molecule is long enough to move this way, its migration speed becomes almost independent of its length. A 200 kb molecule and a 2 Mb molecule both reptate through the gel at essentially the same rate, and they pile up together in a single unresolved band at the top of the gel. A conventional gel is effectively blind to size above 50 kb.
How switching the field restores size separation
PFGE breaks the deadlock by refusing to let the DNA settle into steady end-first motion. Instead of a single constant field, it applies a field that periodically changes direction, typically switching between two directions set at an angle of about 120 degrees to each other.
Each time the field switches, every DNA molecule that had been travelling one way must stop, reorient its long axis, and begin moving in the new direction. This reorientation takes time, and the length of that time depends on size:
A small molecule reorients quickly. A large molecule reorients slowly.
Imagine turning a bicycle around in a corridor versus turning a bus around in the same corridor. The bus spends far longer manoeuvring before it can move off again. In PFGE, while a large fragment is still laboriously realigning itself, a small fragment has already turned and resumed migrating. Every switch of the field therefore costs the large molecule more lost time than the small one.
The interval between switches is called the switch time (or pulse time), and it is the key adjustable parameter. It is chosen to match the size range of interest: short switch times resolve smaller fragments, and progressively longer switch times are needed to resolve larger ones. Many runs ramp the switch time, increasing it gradually through the run, so that a wide range of fragment sizes is resolved on a single gel.
Over many thousands of switches across two to three days, these small per-switch differences accumulate, and the fragments spread out along the gel in order of size, exactly the separation a constant field could never achieve.
The CHEF apparatus
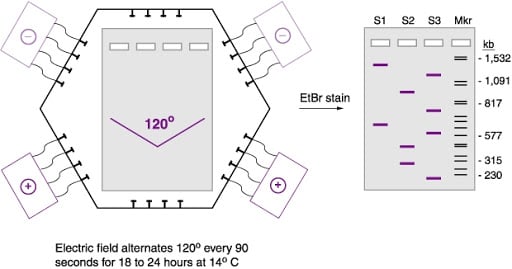
The most widely used PFGE system is CHEF: contour-clamped homogeneous electric field. Its electrodes are arranged in a hexagonal (contour) array around the gel, and they are electronically "clamped" to fixed voltages so that the field is spatially homogeneous: every lane experiences the same field, so all samples run straight and can be compared directly. The field alternates between two directions oriented 120 degrees apart. This homogeneity is what makes CHEF patterns reproducible enough to compare between laboratories, which in turn is what makes a shared database such as PulseNet possible.
Earlier designs, such as the original orthogonal-field (OFAGE) systems, produced curved, distorted lanes that were hard to compare. CHEF solved that problem and became the standard.
Steps of Pulsed-Field Gel Electrophoresis
1. Culture the organism. Streak the isolate (for example Staphylococcus aureus) onto a suitable agar plate and incubate to obtain fresh growth.
2. Embed cells in agarose plugs. Suspend the bacterial cells, mix them with molten agarose, and cast them into plug molds. Embedding is essential: it immobilizes the cells so that the enormous chromosome can be handled without shearing. In free solution, a chromosome this large would fragment from the shear forces of ordinary pipetting, destroying the pattern before it formed.
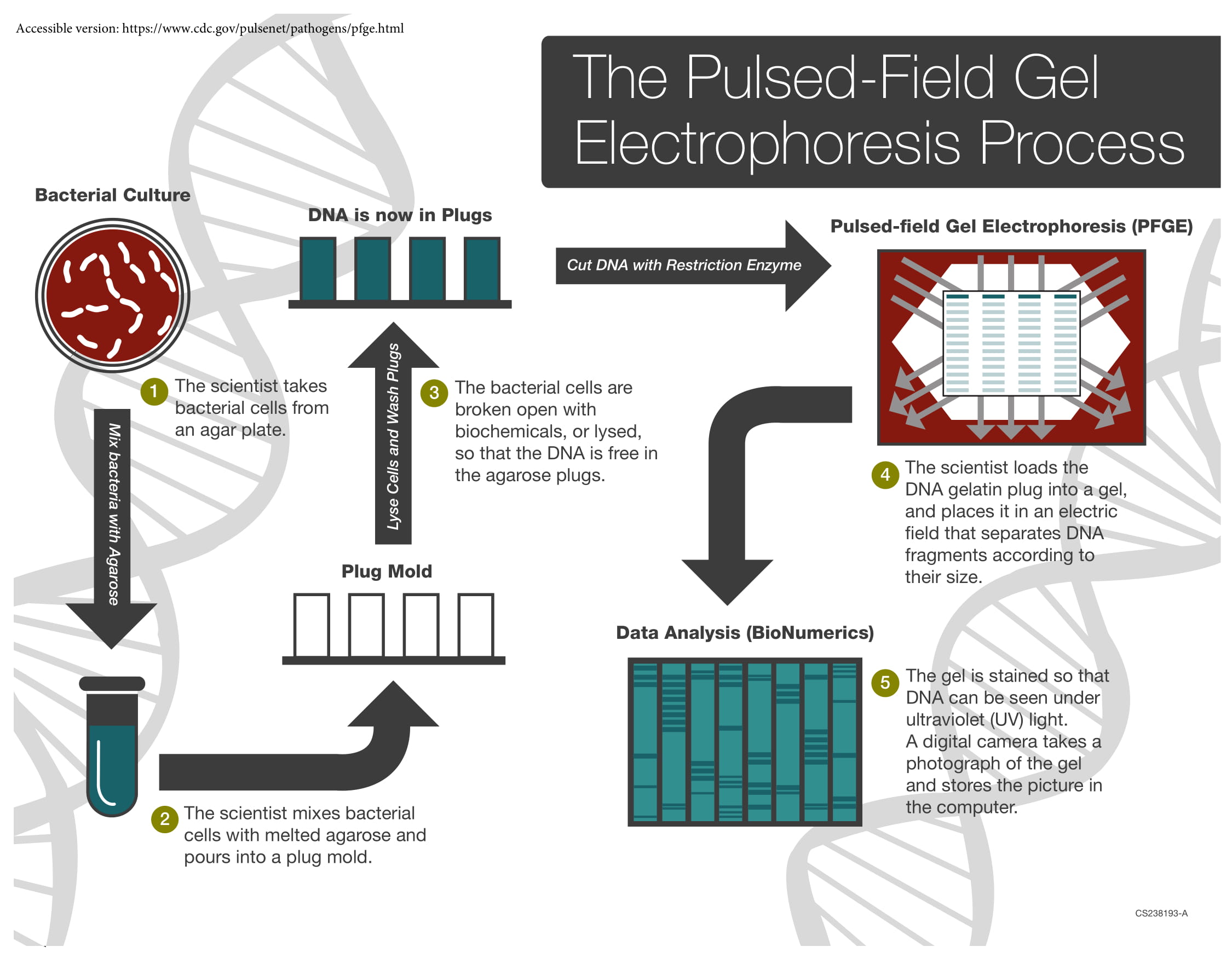 Figure: Steps of Pulsed Field Gel Electrophoresis (Source: CDC)
Figure: Steps of Pulsed Field Gel Electrophoresis (Source: CDC)
3. Lyse the cells in situ. Treat the set plugs with detergents and enzymes to break the cells open and digest proteins, leaving purified, intact genomic DNA held within the agarose. Everything happens inside the plug, so the DNA is never exposed to shear.
4. Digest with a rare-cutting restriction enzyme. Add a restriction enzyme chosen because it recognizes a long (typically 6 to 8 base pair) sequence that occurs only rarely in the genome. Instead of the thousands of fragments a common cutter would produce, the enzyme cuts the chromosome into a manageable number, roughly 10 to 20, of very large fragments. Enzyme choice is organism-specific: XbaI for E. coli and Salmonella, SmaI for S. aureus, AscI for Listeria monocytogenes.
5. Load the plugs and run the pulsed field. Slices of the digested agarose plugs are loaded into the wells of an agarose gel, and separation is driven by an electric field that periodically switches direction between distinct pairs of electrodes.
Each time the field switches, long DNA molecules must reorient before they can migrate in the new direction. The time required to reorient increases with the size of the fragment, so smaller molecules reorient more quickly and larger molecules more slowly. Run for a sufficient time, this reorientation delay separates fragments across a wide size range, from kilobases to megabases, generating a fingerprint profile that can be digitally imaged.
In the widely used CHEF (contour-clamped homogeneous electric field) system, the electrodes are arranged in a hexagonal array and clamped to fixed voltages, so the field is spatially homogeneous and every lane runs straight. The field alternates between two directions oriented about 120° apart, causing the DNA molecules to reorient through that angle at each switch. This homogeneity is what makes patterns reproducible enough to compare between laboratories.
6. Stain and visualize. Stain the gel (ethidium bromide, or a safer alternative) and photograph it under UV using a gel documentation system.
7. Analyze the fingerprint. Normalize the gel image and compare banding patterns using dedicated software such as BioNumerics. Patterns are interpreted for relatedness, often against published criteria, and can be compared across laboratories and against reference databases.
 ### Interpreting PFGE patterns: how related is related?
### Interpreting PFGE patterns: how related is related?
Two isolates rarely produce absolutely identical patterns, so a framework is needed to judge relatedness. The classic Tenover criteria (1995) interpret band differences in the context of a single outbreak:
| Band differences from the outbreak strain | Genetic events | Interpretation |
|---|---|---|
| 0 (indistinguishable) | none | Same strain. Part of the outbreak |
| 2 to 3 bands | a single genetic event | Closely related. Probably part of the outbreak |
| 4 to 6 bands | two independent events | Possibly related. Interpret with caution |
| 7 or more bands | three or more events | Unrelated. Not part of the outbreak |
These thresholds were designed for small, well-defined outbreaks and should not be applied mechanically to large or long-running investigations, but they capture the essential logic: a few band differences can arise from a single mutation or insertion, whereas many band differences imply the isolates are genuinely different strains.
Applications of PFGE
Molecular typing and outbreak investigation. This is the classic use. Because the banding pattern reflects the whole genome, PFGE can distinguish strains that are indistinguishable by phenotypic tests, and can confirm whether isolates from different patients, or from patients and a suspected food or environmental source, are the same strain. It has been central to investigating foodborne outbreaks of Salmonella, Listeria monocytogenes, and Shiga-toxin-producing E. coli.
PulseNet. From 1996, the CDC's PulseNet network standardized PFGE protocols so that public health laboratories across the United States, and later internationally, could upload fingerprints to a shared database and detect outbreaks that span cities or states. Standardization was only possible because CHEF patterns are reproducible between laboratories. PulseNet is the reason PFGE is described as the historical gold standard for foodborne-disease surveillance.
Hospital infection control. PFGE is used to determine whether, for example, several cases of MRSA on a ward represent transmission of one strain or the independent arrival of several, which directly changes the infection-control response.
Bacterial strain characterization and research. It is used to study population structure, genetic diversity, and long-term persistence of strains, and historically underpinned the construction of large-insert genomic libraries.
The move to whole-genome sequencing
PFGE's limitations are real: it takes two to three days, it is technically demanding, it resolves the genome into only a dozen or so bands (so its discriminatory power, while high, is coarse compared with reading the actual sequence), and two isolates can share a PFGE pattern yet differ genetically.
Since roughly the mid-2010s, whole-genome sequencing (WGS) has replaced PFGE for outbreak surveillance in many reference laboratories. PulseNet itself has transitioned to WGS. Sequencing reads the genome base by base, so it offers far higher resolution and can distinguish isolates that PFGE would call identical.
PFGE nonetheless remains relevant. It is still used where sequencing capacity is limited, which includes many laboratories in the settings this site serves, it remains a standard teaching technique, and its enormous historical dataset underpins decades of epidemiology. It is best understood as the technique that made modern molecular epidemiology possible, now being succeeded by sequencing.
How to Remember
Above 50 kb, a normal gel goes blind. Once DNA is far bigger than the gel pores, it stops sieving and starts snaking through end-first, and every large fragment snakes at the same speed regardless of length. 200 kb and 2 Mb land in the same smear. That blindness is the entire problem PFGE exists to solve.
Turn a bicycle, turn a bus. When the field switches direction, every molecule must stop and turn around before it can move on. A small fragment is a bicycle: it pivots and goes. A large fragment is a bus: it spends a long time manoeuvring. The bus loses a little ground at every switch, and after thousands of switches, the buses are far behind the bicycles. That is size separation, rebuilt out of reorientation time.
Switch time is the size dial. Short switch times resolve small fragments; long switch times resolve large ones. Ramp the switch time up through the run and you resolve a wide range on one gel. If you remember one adjustable parameter in PFGE, remember switch time.
Embed first, or you shear. A chromosome this large snaps under ordinary pipetting. So the cells are set into agarose plugs and everything, lysis and digestion, happens inside the plug where the DNA is protected. Embed, lyse in place, cut in place. The DNA never meets a pipette until it is already fragments.
Cut rarely, on purpose. An ordinary restriction enzyme would shred the genome into thousands of pieces and give you a smear. PFGE uses a rare cutter, one that recognizes a long 6-to-8-base site, so the whole chromosome falls into only a dozen or two big pieces: few enough to count, and those counted bands are the fingerprint.
CHEF: Contour-clamped, Homogeneous, so Everyone's Fingerprint is comparable. The homogeneous field means every lane runs straight and identical, so a pattern made in one lab can be matched against a pattern made in another. That reproducibility is the whole basis of PulseNet.
A few bands apart, still family. Many bands apart, strangers. Tenover's rule of thumb: zero band differences means the same strain; two or three means a single mutation away and probably the same outbreak; seven or more means genuinely unrelated. A single genetic event moves only a band or two. Many band differences take many events.
Self-check before you compare two PFGE patterns: Were they cut with the same enzyme, run on the same conditions, in a homogeneous field? Only then can the bands be compared at all.
Key exam facts in one table
| Concept | Fact to retain |
|---|---|
| What PFGE is | Electrophoresis in which the field direction is periodically switched, to separate very large DNA |
| Resolving range | Up to about 10 Mb (largest ever resolved ~14 Mb). Conventional gels stop at ~50 kb |
| Why conventional gels fail above 50 kb | Large DNA moves by reptation (end-first snaking) at a size-independent rate, so fragments co-migrate in one smear |
| Core mechanism | When the field switches, molecules must reorient. Large molecules reorient slowly, small ones quickly, so large ones lose more ground per switch |
| Switch time (pulse time) | The key adjustable parameter. Short for small fragments, long for large. Often ramped during a run |
| Field geometry | Two directions, typically about 120° apart |
| Standard apparatus | CHEF (contour-clamped homogeneous electric field). Hexagonal clamped electrodes give a homogeneous field, so lanes run straight and patterns are comparable between labs |
| Why embed cells in agarose plugs | To prevent shearing of the huge chromosome during lysis and handling |
| Lysis and digestion | Performed in situ, inside the plug |
| Restriction enzyme | A rare cutter (6 to 8 bp recognition site), giving ~10 to 20 large fragments. XbaI (E. coli, Salmonella), SmaI (S. aureus), AscI (Listeria) |
| Run time | Roughly 2 to 3 days total, run of 18-24 h+, with buffer cooling |
| Output | A banding pattern, a whole-genome DNA fingerprint |
| Interpretation | Tenover criteria: 0 bands = same strain; 2-3 = closely related; 4-6 = possibly related; ≥7 = unrelated |
| First described | Schwartz and Cantor, 1984, separating yeast chromosomes |
| Analysis software | BioNumerics and similar |
| Landmark application | PulseNet (CDC, from 1996): standardized PFGE for national and international foodborne-outbreak surveillance |
| Historical status | The gold standard for bacterial molecular typing for ~30 years |
| Current status | Largely superseded by whole-genome sequencing for surveillance (PulseNet has transitioned to WGS); retained where sequencing is unavailable and for teaching |
| Clinical uses | Outbreak investigation (Salmonella, Listeria, STEC); hospital infection control (MRSA transmission); strain characterization |
Where Students Get Confused
"Why can't an ordinary agarose gel just separate large DNA if I run it long enough?" Because time is not the problem; the mechanism is. Below about 50 kb, DNA is sieved by size, and running longer does spread the fragments out. Above 50 kb, the molecule is so much larger than the gel pores that it stops behaving like a coil being sieved and starts snaking through end-first, a motion called reptation. In this regime the migration speed no longer depends on length, so a 200 kb and a 2 Mb fragment travel together no matter how long you run the gel. They are not poorly separated; they are not separated at all.
"How does switching the field direction separate anything?" By turning reorientation into a size test. Every time the field switches, a DNA molecule has to stop and realign its long axis before it can move off in the new direction, and a large molecule takes longer to do this than a small one. During the switch, the small molecule turns quickly and resumes migrating while the large one is still manoeuvring. Each switch costs the large molecule a little more time. Repeat this many thousands of times and the small fragments end up far ahead of the large ones. The separation is built entirely out of the difference in reorientation time.
"What is switch time, and why does it matter so much?" Switch time is the interval between field reversals, and it sets which size range the gel resolves. If the switch time is short, small fragments are resolved and large ones barely separate. If it is long, large fragments separate but small ones run together. Because a real sample spans a wide size range, many runs gradually increase the switch time as the run proceeds, so that different size ranges come into focus at different stages and the whole set is resolved on one gel.
"Why bother embedding the bacteria in agarose plugs? Why not just extract the DNA?" Because a chromosome several megabases long is extremely fragile. The shear forces of ordinary pipetting would snap it into random pieces, and random breakage would destroy the very pattern you are trying to read. Setting the cells into an agarose plug and then lysing and digesting them inside the plug means the DNA is never pipetted as a free, intact molecule. It is only handled once it has already been cut into fragments, which are robust enough to survive.
"Why use an enzyme that cuts rarely? Isn't more cutting better?" The opposite. A common restriction enzyme would cut a bacterial genome in thousands of places and produce an unreadable smear. PFGE deliberately chooses a rare cutter, one whose recognition site is long enough that it occurs only a handful of times in the whole genome, so the chromosome falls into perhaps ten to twenty very large pieces. Few, large, countable bands are what make a comparable fingerprint. Many small ones would not.
"Two isolates have the same PFGE pattern. Are they definitely the same strain?" Not definitely. PFGE reads the genome as only a dozen or two bands, so it is possible for two genuinely different isolates to share a pattern by chance, or for outbreak-related isolates to differ by a band or two through a single mutation. The Tenover criteria exist precisely to interpret small differences: zero band differences suggests the same strain, a few differences suggests close relatedness, and seven or more suggests the isolates are unrelated. This coarseness is exactly why whole-genome sequencing, which reads every base, has overtaken PFGE for surveillance.
"Is PFGE still used, or has sequencing replaced it?" Both are true. In well-resourced reference laboratories, whole-genome sequencing has largely replaced PFGE for outbreak surveillance, and PulseNet itself has moved to sequencing. But PFGE is still used where sequencing capacity is limited, it remains a standard technique to learn, and thirty years of PFGE data underpin the epidemiology that preceded the sequencing era. It is the foundation the current methods were built on, not a discarded curiosity.
References and further readings
- Schwartz DC, Cantor CR. Separation of yeast chromosome-sized DNAs by pulsed field gradient gel electrophoresis. Cell. 1984;37(1):67-75. doi:10.1016/0092-8674(84)90301-5
- Tenover FC, Arbeit RD, Goering RV, Mickelsen PA, Murray BE, Persing DH, et al. Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. Journal of Clinical Microbiology. 1995;33(9):2233-2239.
- Centers for Disease Control and Prevention. PulseNet: Pulsed-field Gel Electrophoresis (PFGE). Atlanta: CDC. Available from: https://www.cdc.gov/pulsenet/pathogens/pfge.html
- Sharma-Kuinkel BK, Rude TH, Fowler VG Jr. Pulse field gel electrophoresis. Methods in Molecular Biology. 2016;1373:117-130.
- Ribot EM, Freeman M, Hise KB, Gerner-Smidt P. PulseNet: entering the age of next-generation sequencing. Foodborne Pathogens and Disease. 2019;16(7):451-456.

Tankeshwar Acharya, MSc (Medical Microbiology)
Tankeshwar Acharya is an Assistant Professor in the Department of Microbiology at Patan Academy of Health Sciences (PAHS), Nepal, where he has been teaching and practicing clinical microbiology for over 14 years. He is the founder of Microbe Online, one of the leading free microbiology education resources on the web, covering bacteriology, mycology, parasitology, immunology, and clinical laboratory diagnostics written from direct experience in both the classroom and the diagnostic laboratory.